Митохондриальная дисфункция при болезни Фридрейха: биохимические и цитохимические аспекты
Ершова М.В., Иллариошкин С.Н., Сухоруков B.C., Клюшников С.А., Федорова Т.Н., Иванова-Смоленская И.Л.
НИИ неврологии РАМН, г.Москва
НИИ педиатрии и детской хирургии Минздрава России, г.Москва
Реферат. Проанализированы биохимические и цитохимические показатели у больных с ДНК-подтвержденным диагнозом болезни Фридрейха. Выявлены нарушения в системе окислительного фосфорилирования митохондрий при болезни Фридрейха, а также их взаимосвязь с характером течения заболевания при ее различных клинических вариантах. Прослежена отчетливая тенденция к нормализации этих цитохимических показателей при проведении однократного курса лечения препаратами, стабилизирующими функции митохондрий.
Болезнь (атаксия) Фридрейха (БФ) - тяжелое нейродегенеративное аутосомно-рецессивное заболевание, представляющее собой наиболее часто встречающуюся форму наследственных атаксий. Распространенность БФ составляет 1 на 50000 населения, частота гетерозиготного носительства мутантного гена - 1 на 90-120 человек [11, 13, 15, 16].
До последнего времени БФ считалась отчетливо выраженной нозологической формой, имеющей достаточно строгие и общепринятые диагностические критерии [13, 15]: 1) аутосомно-рецессивное наследование; 2) начало заболевания до 25 лет; 3) прогрессирующая атаксия; 4) дизартрия; 5) сухожильная арефлексия; 6) симптом Бабинского; 7) утрата глубокой чувствительности в дистальных отделах конечностей; 8) электрофизиологические признаки аксональной сенсорной невропатии (в пределах 5 лет от начала заболевания); 9) изменения на ЭКГ. Патоморфологически БФ характеризуется гибелью афферентных волокон задних корешков спинного мозга и периферических нервов, а также комбинированной дегенерацией чувствительных ганглиев, задних и боковых столбов спинного мозга с утратой больших чувствительных нейронов, изменениями в пирамидном и спиномозжечковых путях [16].
Предположения о роли митохондриальных нарушений в патогенезе БФ высказывались еще в 80-е годы XX века A.Barbeau и его школой [4]. Они основывались на том, что классическая клиническая картина БФ складывается из своеобразной комбинации симптомов, которая обычно свойственна митохондриальным энцефаломиопатиям (поражение ЦНС, сердечной мышцы, поджелудочной железы, органа зрения ит.д.). Новый этап в исследовании патогенетических основ БФ наступил в середине 1990-х годов, когда ген заболевания Х25 (FRDA, индекс по каталогу MIM-229300) был идентифицирован на хромосоме 9ql3 [7]. Этот ген состоит из 7 экзонов и кодирует белок, состоящий из 210 аминокислот и получивший название фратаксин [6]. При БФ имеет место экспансия тандемных тринуклеотидных повторов гуанин-аденин-аденин (GAA), расположенных в 1-м интроне гена Х25: в норме число GAA-повторов не превышает 36, тогда как у 96-98% пациентов с БФ варьирует от 66 до 1700 копий и более [6]. Аномально удлиненный участок гена нарушает созревание первичного транскрипта, препятствуя вырезанию интрона из молекулы зрелой мРНК, что приводит к пропорциональному снижению или полному блоку трансляции с недостаточностью (отсутствием) в тканях нормального белка фратаксина. Кроме того, эксперименты с использованием экспрессионных систем in vitro и in vivo показали, что в результате мутации изменяется конформация мутировавшего участка ДНК, соответственно возникают угнетение экспрессии гена и дефект синтеза фратаксина [21]. Около 2-4% пациентов, страдающих БФ, являются компаунд-гетерозиготами по мутациям в гене фратаксина: у таких больных в одном аллеле гена имеет место экспансия тринуклеотидных GAA-повторов, а в другом - точковая мутация с заменой нуклеотида. Точковые мутации локализуются в кодирующей части гена фратаксина и ведут к синтезу функционально дефектного белка [5, 8].
Результаты исследований последних лет свидетельствуют о ключевой роли белка фратаксина в регуляции митохондриального транспорта железа [14, 19-21]. Нарушение функции фратаксина в результате мутаций в гене Х25 сопровождается повышением содержания железа в митохондриях, снижением резистентности к оксидантному стрессу и окислительным повреждением митохондрий, снижением синтеза АТФ и нарушением энергетического метаболизма клетки [18, 20, 24-26]. Важное значение в патогенезе болезни придается также развивающейся недостаточности Fe-S-зависимых субъединиц ферментов дыхательной цепи митохондрий [19, 20-23]. С современных позиций БФ рассматривается как особая разновидность митохондриальной болезни, обусловленная повреждением ядерного гена.
Таким образом, в связи с ключевой ролью поражения митохондриального белка фратаксина в патогенезе БФ и необходимостью адекватного биохимического мониторинга за состоянием больных, а также в связи с принципиальной возможностью проведения им терапии препаратами, улучшающими деятельность митохондрий, в настоящее время весьма актуальна разработка информативных методов исследования митохондриальной функции у пациентов с БФ.
Целью настоящей работы являлся анализ нарушений в системе окислительного фосфорилирования митохондрий у пациентов с БФ, а также их взаимосвязи с характером течения заболевания при различных клинических вариантах БФ. Поскольку главным признаком митохондриальной недостаточности служит нарушение активности окислительно-восстановительных ферментов, их исследование было выбрано в качестве основы для данной работы.
Нами была обследована сплошная невыборочная серия больных молодого возраста с различными клиническими вариантами дегенеративных атаксий (70 чел.), направленных в нейрогенетическое отделение НИИ неврологии РАМН из различных медицинских учреждений для уточнения диагноза.
Обследование больных включало, помимо детального неврологического осмотра и традиционных общеклинических тестов, использование различных современных лабораторных и инструментальных методов исследования (электронейромиография, электро-и эхокардиография; определение содержания молочной, пировиноградной кислот и их соотношения в периферической крови; изучение мультимодальных вызванных потенциалов, компьютерная рентгеновская и магнитно-резонансная томография головного и спинного мозга). Клинический статус исследован по модифицированной шкале Pourcher и Barbeau (1978).
В результате ДНК-диагностики было выявлено 23 пациента с подтвержденным диагнозом БФ. Больные находились под наблюдением в отделении на протяжении различных периодов времени (от 3 мес до 7 лет). Средний возраст пациентов с БФ составлял 24,4 ± 12,5 года (от 7 до 59 лет). Заболевание впервые проявилось в среднем в 13,8 ± 9,7 года (от 5 до 46 лет). Контрольная выборка была сформирована из 28 практически здоровых людей возрастной группы, сходной с группой выявленных пациентов с БФ.
Пациенты молодого возраста, у которых при наличии сходной с БФ клинической картины заболевания имелся отрицательный результат ДНК-диагностики (исключены мутации в гене фратаксина), были взяты в качестве группы сравнения. Кроме того, также в качестве дополнительной группы сравнения были обследованы 12 пациентов с аутосомно-доминантными формами спиноцеребеллярных атаксий (СЦА) 1, 2, 3 и 6-го типов.
Молекулярно-генетическое исследование включало амплификацию области гена Х25, содержащей тандемные тринуклеотидные повторы с использованием стандартных праймеров [12] и модифицированных условий полимеразной цепной реакции (ПЦР). Полученные в результате реакции продукты амплификации окрашивали бромидом этидия и визуализировали в 0,9% агарозном геле.
Системная оценка энергетического метаболизма у обследованных больных проводилась путем определения активности ферментов тканевого дыхания - сукцинатдегидрогеназы (СДГ), а-глицерофосфатдегидрогеназы (ГФДГ), глутаматдегидрогеназы(ГДГ), лактатдегидрогеназы (ЛДГ) и малатдегидрогеназы (МДГ) - в лимфоцитах периферической крови с использованием цитохимического метода Р.П. Нарциссова [2, 3]. При использовании данного метода ферментативная активность выражается в условных единицах, соответствующих среднему числу гранул формазана, являющегося продуктом цитохимической реакции. У 7 больных БФ, помимо этого, был проанализирован ферментный статус лимфоцитов с помощью метода компьютерной телеметрии с использованием диагностической системы, состоящей из персонального компьютера высокой мощности со специальным пакетом морфо-метрических программ "Видеотест Авто 4.0" (Санкт-Петербург) и светового микроскопа с цифровой видеокамерой. Оценивали следующие параметры: количество и площадь депозитов формазана, интервал яркости, оптическую плотность, эллипс и др. У 3 пациентов с БФ были исследованы мышечные биоптаты (четырехглавая мышца бедра) с помощью световой и электронной микроскопии стандартными методами, а также путем цитохимической оценки активности СДГ (по Нахласу) и цитохромоксидазы (по Берстону). Состояние перекисного окисления липидов (ПОЛ) изучали по наиболее информативным параметрам Ре2+-индуцированной хемилюминесценции липопротеинов сыворотки крови: 1) уровень первичных продуктов ПОЛ (преимущественно гидроперекисей); 2) способность липопротеиновых структур к ПОЛ; 3) состояние антиоксидантной системы крови.
Статистический анализ полученных данных проводился с использованием пакета программ STATISTICA (StatSoft Inc. 1993 USA).
Результаты молекулярно-генетического исследования и клиника-генетические сопоставления. Среди 70 больных молодого возраста с прогрессирующей спорадической или аутосомно-рецессивной атаксией, фенотипически сходной с БФ, при проведении прямой ДНК-диагностики гомозиготная мутация в гене Х25 была выявлена у 23 человек. Таким образом, доля случаев БФ при мутационном скрининге гена Х25 в нашей выборке больных составила 33%. Тяжесть мутации (количество тандемных тринуклеотидных GAA-повторов) варьировала от 200 до 1100 копий триплетов. Нами выявлена обратная корреляция между числом GAA-повторов и возрастом дебюта заболевания у обследованных больных (R=0,67; р=0,0008), что согласуется с аналогичными данными в отечественной и зарубежной литературе [1, 6, 9, 10, 12, 17, 20]. Среди обследованных больных классическая картина заболевания наблюдалась у большинства пациентов. Разнообразные атипичные формы, представляющие большую сложность с точки зрения клинической диагностики БФ, были отмечены в 26,1% случаев (табл. 1). Частота экстраневральных проявлений болезни, свидетельствующих о наличии клинически значимого генерализованного биохимического дефекта, представлена в табл. 2.
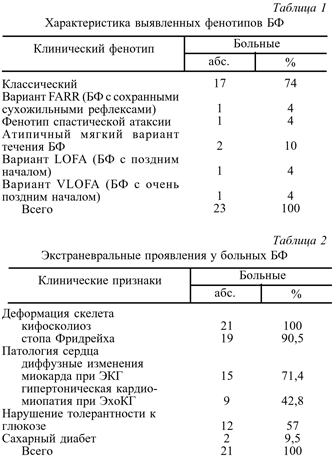
Основные биохимические признаки нарушения клеточной энергетики.При изучении основных биохимических показателей энергообмена (глюкоза крови, сахарная кривая, уровни молочной и пировиноградной кислот, их соотношения) у пациентов с БФ были выявлены общепризнанные маркеры нарушения функционирования митохондрий. Уровень глюкозы в крови натощак был в пределах нормы у всех больных, кроме 2 пациентов с развернутой клинической картиной сахарного диабета. В то же время у 12 (57%) из 21 больного отмечались изменения на сахарной кривой, а также значительная гиперлактат- и гиперпируватемия как до, так и после нагрузочной пробы с глюкозой.
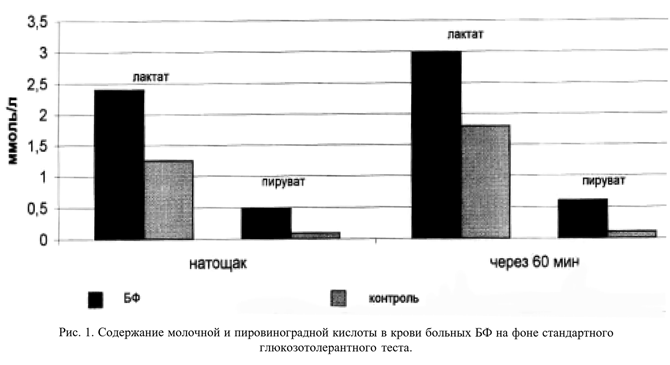
У больных БФ были выявлены прямые признаки системного лактат-ацидоза (рис 1): уровень молочной и пировиноградной кислот натощак у больных БФ составил соответственно 2,4±0,9 и 0,5±0,3 ммоль/л (норма-соответственно до 1,3 и до 0,1 ммоль/л). После приема глюкозы (через 60 мин) уровень молочной (3,0±0,7 ммоль/л) и пировиноградной(0,6±0,3 ммоль/л) кислоты оставался достоверно выше, чем в контроле (норма-соответственно до 1,8 и до 0,1 ммоль/л). Коэффициент лактат/пируват был повышен (значение Л/П >10) у 4 больных, что свидетельствовало о нарушении окислительного фосфорилирования в клетках и являлось важным биохимическим маркером дефицита синтеза АТФ, сопровождающегося ацидификацией тканей.
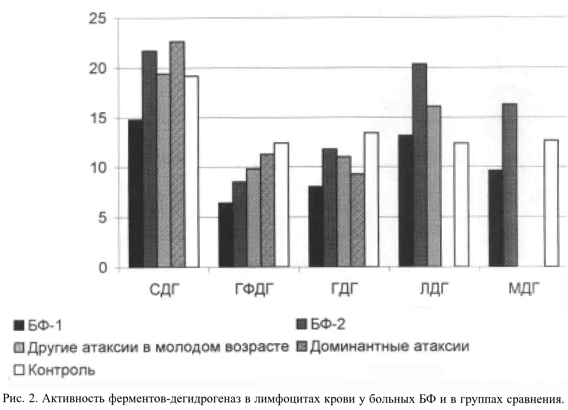
У всех больных БФ были обнаружены значительные нарушения в системе перекисного окисления липидов, выражающиеся преимущественно в достоверном снижении активности собственной антиоксидантной системы крови, оцениваемой по τ-параметру. Этот показатель у больных БФ составил 34,1± 18,5 с при контрольных значениях τ не ниже 90 с (р=0,02). Примечательно, что у подавляющего числа больных БФ (у 21 из 23) не было установлено повышение уровня первичных предобразованных продуктов ПОЛ. Способность липопротеиновых структур к перекисному окислению также были повышены только у 2 из 23 больных. Таким образом, полученные нами данные свидетельствуют о том, что окислительный стресс при БФ связан с нарушениями в системе антиоксидантной защиты. Этот вывод имеет важное теоретическое и практическое значение, демонстрируя один из возможных механизмов реализации дефекта фратаксина в тканях и создавая предпосылки для разработки патогенетических методов лечения БФ. Цитохимический анализ функции митохондрий. Ферментативный статус лимфоцитов был исследован у 21 больного БФ и у всех больных из групп сравнения, причем у пациентов с БФ - дважды: до и после проведения однократного курса стационарного лечения препаратами, поддерживающими функцию митохондрий. При оценке активности СДГ лимфоцитов в группе БФ у пациентов были выявлены разнонаправленные изменения, что позволило разделить эту группу на 2 подгруппы: БФ-1 - пациенты с пониженной активностью дегидрогеназ (n=15, т.е. большинство больных) и БФ-2 - с повышенной активностью СДГ, ЛДГ и МДГ (n=6). Общие результаты цитохимического анализа представлены в табл. 3. Достоверное снижение активности ключевого митохондриального фермента СДГ, выявленное нами у 71% больных БФ, свидетельствует о выраженном системном нарушении биоэнергетических процессов при данном заболевании.
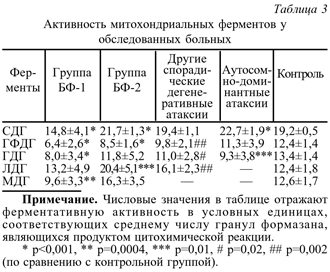
Примечательно, что в группе сравнения активность СДГ не отличалась от значений в контрольной группе. Это указывает на отсутствие системных нарушений клеточной энергетики при спорадических атаксиях в молодом возрасте и на то, что патогенетические механизмы развития этих заболеваний значительно отличаются от генеза БФ. При анализе данных не было получено корреляций между числом GAA-повторов (т.е. степенью генетического дефекта), тяжестью состояния по клинической шкале, продолжительностью заболевания и степенью снижения активности СДГ, что свидетельствует о сложности и многоступенчатости биохимических нарушений, приводящих к системному дефекту метаболизма. Активность большинства других исследованных ферментов (ГФДГ, ГДГ, МДГ) в подгруппе БФ-1 была также достоверно ниже, чем в контроле (см. рис. 2 и табл. 3).
Особого внимания заслуживает факт достоверного повышения средней активности СДГ по сравнению с контролем у части больных БФ. Это может быть расценено как мобилизация энергетических ресурсов благодаря ускорению окисления сукцинатов. Очевидно, однако, что в такой ситуации деятельность организма протекает при полной мобилизации функционального резерва, что в конечном счете может привести к истощению метаболических функций. Аналогичная закономерность в данной подгруппе больных наблюдалась и для других ферментов (см. табл. 3 и рис. 2), причем уровень ЛДГ и МДГ в группе БФ-2 оказался выше контрольных цифр (р=0,01). Данная группа пациентов отличалась от основной группы пациентов с БФ более высоким числом GAA-повторов (р=0,03), ранним дебютом заболевания (р=0,04), высоким уровнем лактат-ацидоза в крови (р=0,05) и более низким уровнем собственной антиоксидантной защиты (р=0,02).
При проведении компьютерной телеметрии при БФ были выявлены следующие особенности данных морфометрического анализа:
1. У пациентов с БФ по сравнению с контролем имело место уменьшение числа гранул продукта реакции, более низкая оптическая плотность депозитов и увеличение значения интегральной оптической плотности (р=0,04), что достоверно свидетельствовало о патологии энергетического метаболизма в митохондриях.
2. В процентном отношении у пациентов с БФ отмечалось более высокое содержание кластеров (кластеризация гранул) по сравнению с контролем, что является признаком напряжения энергетического метаболизма (рис. 3). Прослеживалась тенденция к увеличению площади кластеров с нарастанием тяжести заболевания в клинической картине. В контрольной группе кластеризация гранул была выражена значительно меньше, что свидетельствовало о покое клетки в условиях эффективного и достаточного окислительного фосфорилирования.
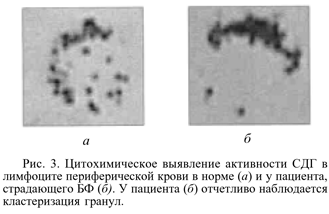
3. Выявлено уменьшение количества мелких гранул и значения эллипса в исследованных клетках по сравнению с контролем, что является признаком дисбаланса митохондриального метаболизма.
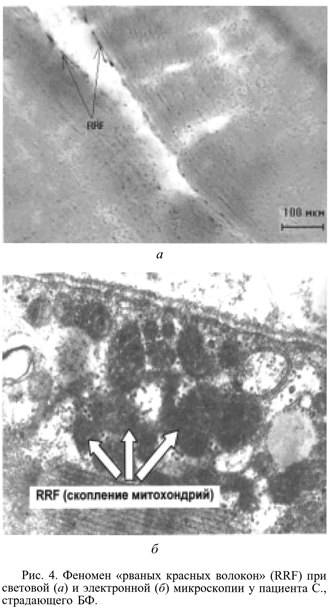
Анализ биоптатов скелетных мышц. Для подтверждения митохондриальных нарушений на морфологическом уровне были исследованы инцизионные биоптаты четырехглавой мышцы бедра у 3 пациентов с классическим фенотипом БФ, включавшим гипертрофическую кардиомиопатию. У всех больных были обнаружены признаки митохондриальной патологии, особенно выраженные у одного из них (рис. 4): при световой микроскопии выявлялись умеренная активность СДГ и ЦО-позитивный феномен рваных красных волокон (RRF) в 7% миоцитов (норма до 5%), а также субсарколеммальные скопления гликогена, липидов и кальция. У 2 других пациентов умеренные, но отчетливые признаки патологии митохондрий определялись только при электронной микроскопии (снижение количества митохондрий, вакуолизация матрикса, дегенерация крист). Полученные нами результаты морфологического исследования мышц у пациентов с БФ являются важным доказательством митохондриальной природы заболевания.
Динамика морфометрических показателей ферментативной активности на фоне терапии у больных БФ. Повторное цитохимическое исследование активности дегидрогеназ в группе больных БФ проводилось после трехнедельного курса лечения, направленного на повышение эффективности биологических процессов тканевого дыхания, окислительного фосфорилирования и продукции макроэргических фосфатов. Терапия включала препараты, повышающие активность дыхательной цепи митохондрий (коэнзим Q10, янтарная кислота, цитохром С и др.), кофакторы энзимных реакций энергетического обмена (рибофлавин, тиамин, липоевая кислота, карнитин и др.), препараты, уменьшающие лактат-ацидоз, а также антиоксиданты. Нами была прослежена следующая динамика цитохимических и клинических показателей: а) в группе БФ-2 с исходно повышенной активностью СДГ данный ключевой параметр снижался, а в группе БФ-1 - имел отчетливую тенденцию к повышению (вплоть до нормальных значений); б) отмечена положительная динамика таких клинических показателей, как повышение толерантности к физическим нагрузкам, снижение утомляемости, усиление двигательной активности, увеличение мышечной силы.
Таким образом, полученные нами результаты могут быть первым шагом на пути к разработке патогенетически обоснованной схемы поддерживающей терапии больных БФ. Длительность эффекта однократного курса лечения, необходимая частота повторных курсов или продолжительность поддерживающей терапии у пациентов с БФ входит в задачу последующих исследований.
Литература
1. Иллариошкин С.Н., Друзина Е.Б., Багиева Г.Х. и др. // Журн. неврол. и психиатр, им. С.С. Корсакова. - 1999. - №8.- С.31-34.
2. Нарциссов Р.П. // Педиатрия. - 1998 - № 4. - С.101-105.
3. Нарциссов Р.П. // Арх. анатом, гистол. и эмбриол. - 1969. - № 5. - С.85-91.
4. Barhean А. // Can. J. Neurol. Sci. - 1984. - Vol. 11. - P.646-660.
5. Bidichandani S.I., Ashizawa T, Patel P.I. // Am. J. Hum. Genet- 1997. - Vol. 60. - P.1251-1256.
6. Campuzano V., Montermini L., Koutnikova H. et al. // Science. - 1996. - Vol. 271. - P.1423-1427.
7. Chamberlain S., Shaw J., Rowland A.., Wallis J. et al. // Nature. - 1988. - Vol. 334. - P.248-250.
8. Cossee M., Durr A.., Cavalcanti F. et al. // Ann. Neurol. -1999. - Vol. 45. - P.200-206.
9. Dnizina E., Illarioshkin S., Ovchinnikov I. et al. // Med. Genet. - 1997. - Vol. 9 (SuppL). - P.50.
10. Durr A., Cossee M., Agid Y. et al. // N. Engl. J. Med. - 1996.- Vol. 335.- P.1169-1175.
11. Filla A.,De Michele G. // J. Neurol. - 1992. - Vol. 239. - P.351-353.
12. Pitta A., De Michele G., Cavalcanti F. et al. // Am. J. Hum. Genet. - 1996. - Vol. 59. - P.554-560.
13. Geoffrey G., Barbeau A., Breton G. et al. // Can. J. Neurol. Sci. - 1976. - Vol. 3. - P.279-286.
14. GrayJ. V., Johnson K.J. // Nat. Gen. - 1997. - Vol. 16. - P.323-328.
15. Harding A.E. // Brain. - 1981. - Vol. 104. - P.598-620.
16. Harding A.E. The hereditary ataxias and related disorders. Edinburgh: Churchill Livingstone, 1984.
17. Wariosbkm S.N., Bagieva G.Kh, Klyushnikov S.A. et al. // Eur. J. Neurol. - 2000. - Vol. 7. - P.535-540.
18. Lodi R., Cooper J., Bradley J. et al. // Proc. Nat. Acad. Sci. USA. - 1999. - Vol. 96. - P.11492-11495.
19. PandolfoM. // Mov. Disord. - 2001. - Vol. 16. - P.815-821.
20. Pandolfo M. // Neuromus Dis. -1998. - Vol. 8. - P.409-415.
21. Puccio H, KoenigM.//I Hum. Mol. Gen. 2000. - Vol. 9. - P. 887-892.
22. Rotig A., De Lonlay P., Chretien D. et al. // Nat. Genet. 1997. - Vol. 17. - P.215-217.
23. Rotig A., Lonlay P., Chretien D. et al. // Nat. Genet. - 1997. - Vol. 17. - P.215-218.
24. Schniz J.B., Dehmer Т., Schols L. et al. // Neurology. - 2000. - Vol. 55. - P.1719-1721.
25. Sherer Т., Greenamyre J.T. // Neurology. - 2000. - Vol. 55.-P.1601-1602.
26. Vorgerd M., Schols L, Hardt C. et al. // Neuromusc. Disord. - 2000. - Vol. 10. - P.430-435.
Ершова М.В., Иллариошкин С.Н., Сухоруков B.C., Клюшников С.А., Федорова Т.Н., Иванова-Смоленская И.Л. Митохондриальная дисфункция при болезни Фридрейха: биохимические и цитохимические аспекты // Неврологический вестник. - 2002. - Т. XXXIV, вып. 3-4. - С.5-11.